
Over the last 4 years we have developed a better way to bioengineer organisms. We are now open sourcing our entire software stack, 20n/act. Find it at https://github.com/20n/act.
The stack will enumerate all bio-accessible chemicals, called reachables (20n/act/reachables). For each of those chemicals, it will design DNA blueprints. These DNA blueprints can bioengineering organisms with un-natural function. E.g., build organisms to make chemicals that were previously only sourced through petrochemistry.
To do that, the stack contains many modules built from scratch in-house. Some of them: mine raw biochemical data, integrate heterogenous sources, learn rules of biochemistry, automatically clean bad data, mine patents, mine plain text, bioinformatic identification of enzymes with desired function.
Once the suggested DNA is used to create new engineered cells those cells can be analyzed with LCMS for function. Our deep learning-based untargeted metabolomics stack processes the raw data and enumerates all side-effects of the changed genomic structure of the cell. Some would be expected, as the organism making the desired chemical, and some unexpected metabolic changes are highlighted.
We are also releasing a economic cost model for bioproduction. This economic cost model maps the desired the market price of the biological product to the "science needed" to get there. The "science needed" is measured in fermentation metrics, yield, titers, productivities of the engineered organisms. From that one can draw estimates of cost of investment (time and money) needed, and the expected ROI. More on this in a later blog post.
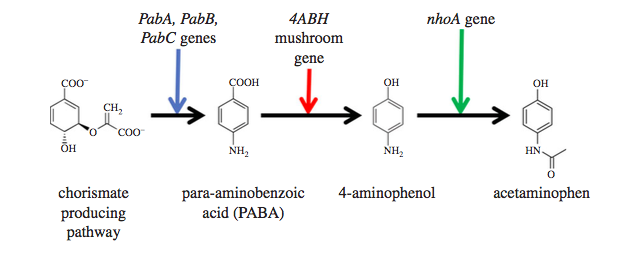
Our Bioreachables Service predicted that we could make acetaminophen biologically. A world's first. Acetaminophen/Paracetamol is the active ingredient of the widely used analgesic Tylenol™. There is no other biological process on the planet that can make acetaminophen (APAP) from sugars. The entire supply of our APAP comes from petroleum through one of two major processes. Using our software predictions, we engineered an E. coli and a yeast (S. cerevisiae) with a gene from a mushroom (4ABH), and over-expressed two native genes (PabABC, and nhoA). When grown with sugar, this yeast will ferment those sugars into APAP. In the image below, the vials from left to right contain, sugar → broth from our engineered yeast → clarified fermentation supernatant → separated APAP powder.

The system suggested the pathway below. A pathway is an ordered list of intermediate chemicals, with each step being catalyzed by a new or over-expressed gene. APAP's predicted pathway starts at chemicals already present in the cell, and uses the extra genes we insert to convert it one step at a time to the final product. The pathway came entirely through the software predictions, with no human input. In fact, we were not even asking it for a pathway to acetaminophen. Our Bioreachables Service enumerates all possible bio-molecules, i.e., molecules that can be made biologically through gene insertions into cells. APAP was conspicuous. Even since its introduction in 1955, it has been sourced from petrochemical synthesis. No natural cell, including plants and other species, make it and yet it appeared to be in the purview of enzymatic chemistry. Or at least, so our software predictions indicated. We did the experiment as suggested and confirmed production.
We are happy to provide the LCMS dataset for APAP confirmation. Please email to request access. The LCMS data is large, and we use our deep learning LCMS analytics to inspect it comprehensively, and there are public software modules that can process pieces of it. Contact us too if you are interested in the comprehensive analysis.
Biology faces a debugging hurdle. Biochemical systems (cells) are complex, and inspecting them is non-trivial. Observing chemicals you know about is difficult; even harder is seeing unexpected chemical changes. This debugging challenge, colloquially a known unknown and unknown unknown problem, complicates forward progress. Given that it is possible to collect incredible amounts of multi-dimensional, dense biological data, we wondered if deep learning can help debug biology.
Over the last two years, we have engineered 180+ yeast and bacterial cells to produce industrially relevant chemicals, e.g., one of our yeasts is the first cell that makes acetaminophen. We recently started looking at human diseased cells as well. Be it engineered microbial cells or human diseased cells, both are non-trivial to debug, i.e., identifying differences between modified cells vs. controls. Let's call normal cells (wild type or healthy cells) the controls, and the alternative cells the variant (modified through genetic manipulation, or human diseased lines.)
State-of-the-art: Scientists pick one or a few chemicals out of possibly millions, and check if the variant differs from the controls for each of those chemicals. They do this "diff" by comparing the data trace from an analytic instrument, such as LCMS. Typical workflows have tens, if not hundreds or thousands, of false negatives, and due to confirmation biases can have false positives as well. This is complicated by the fact that LCMS instruments have finite precision and there are collisions between signals of different chemicals.
Deep learnt LCMS analysis: We input raw LCMS data (without biases towards individual conjectured molecules) and ask our deep learnt model to evaluate which molecules are different in the variant samples.
Take for example a yeast we recently constructed to produce acetaminophen. It was a novel pathway built using predictions on new gene insertions, with a few genes knocked out. We grew these variant strains on glucose media and concurrently grew wild type baker's yeast as controls. We put that through our pipeline, and it outputted a ranked list of ~200 molecules. Rank 5 on the list is the following peak (on the left is the variant, and the right is the control):
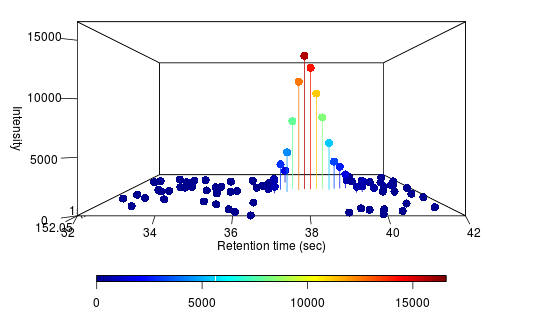
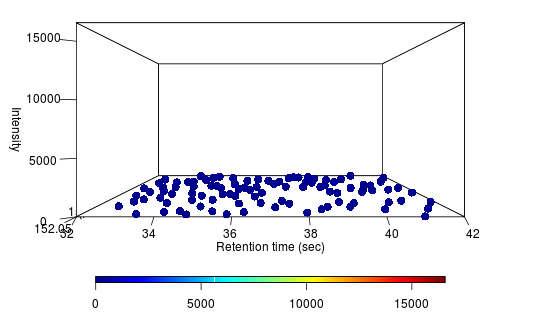
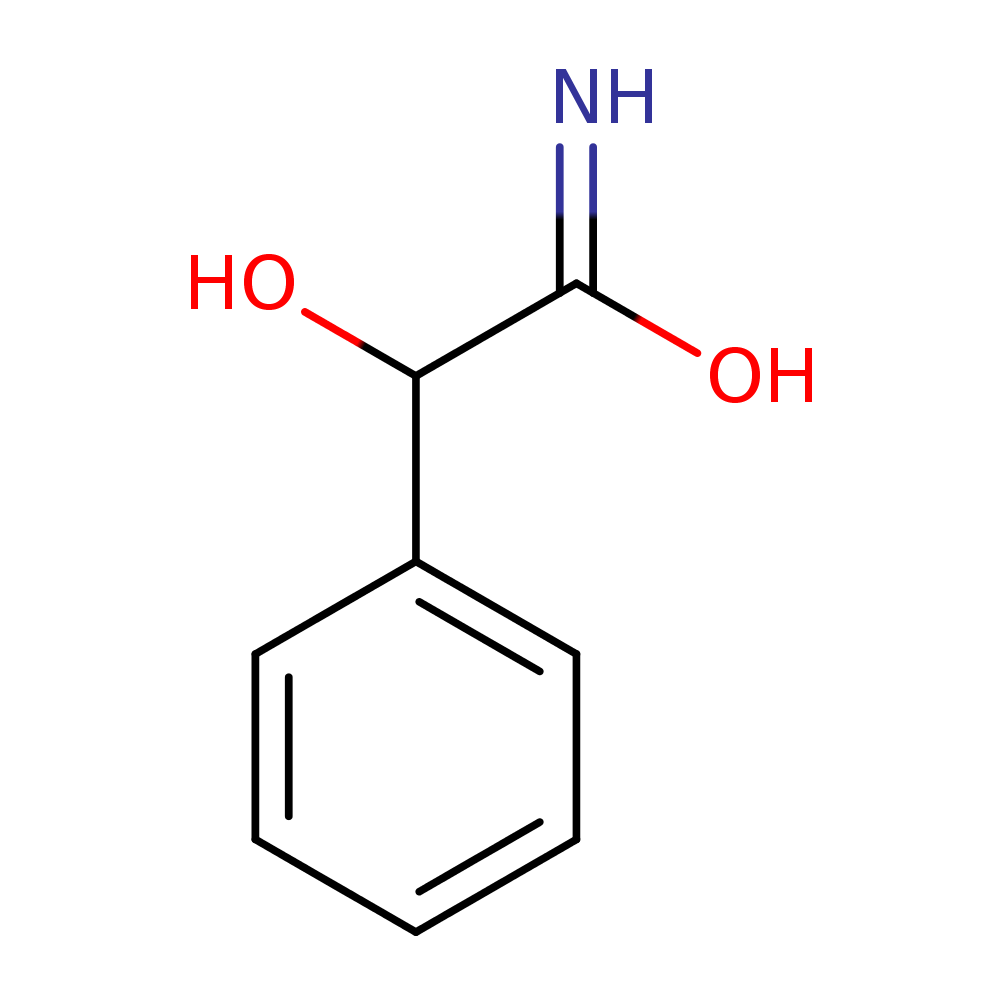
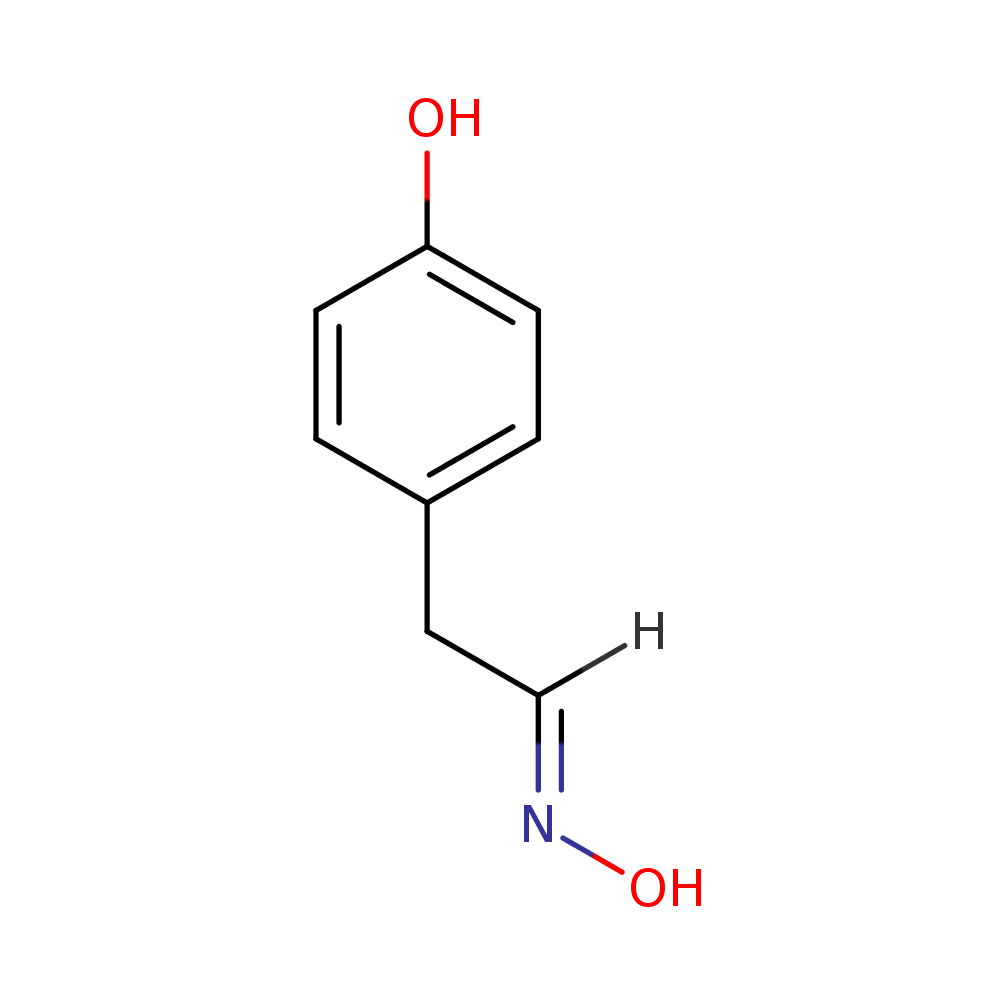
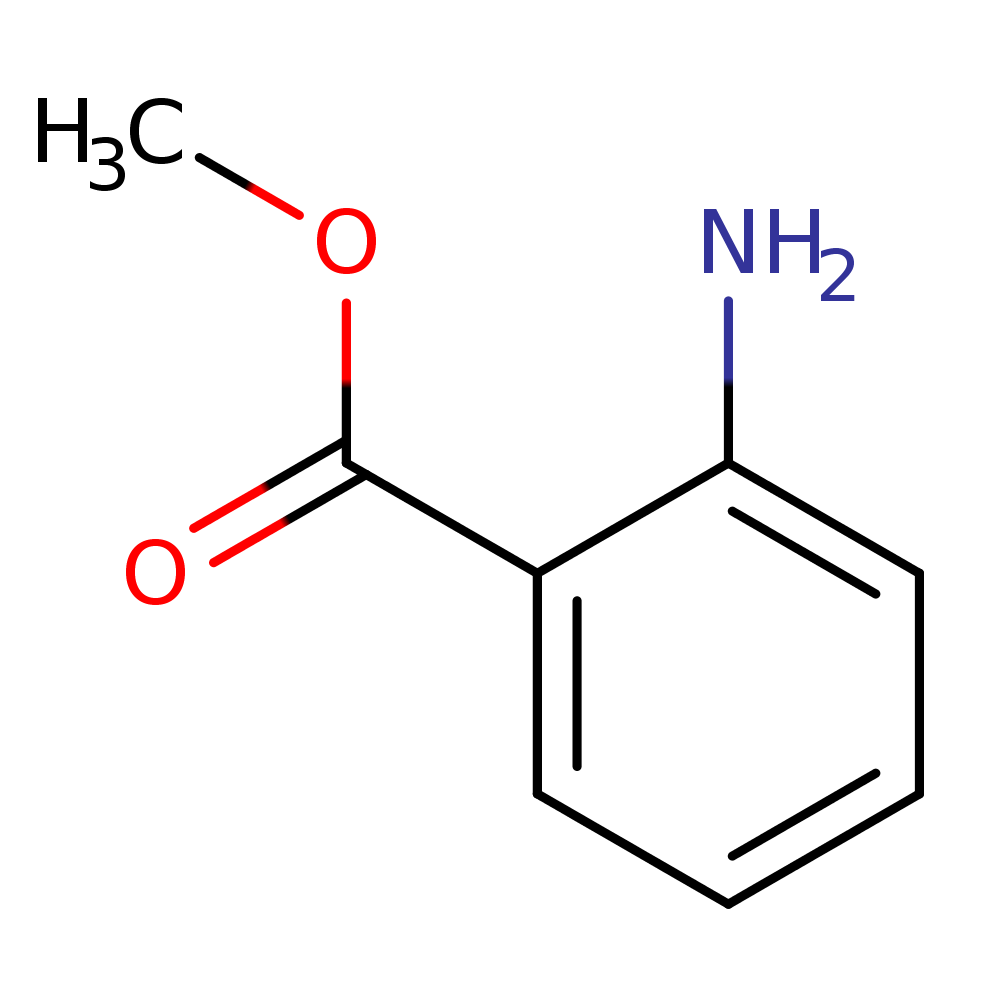
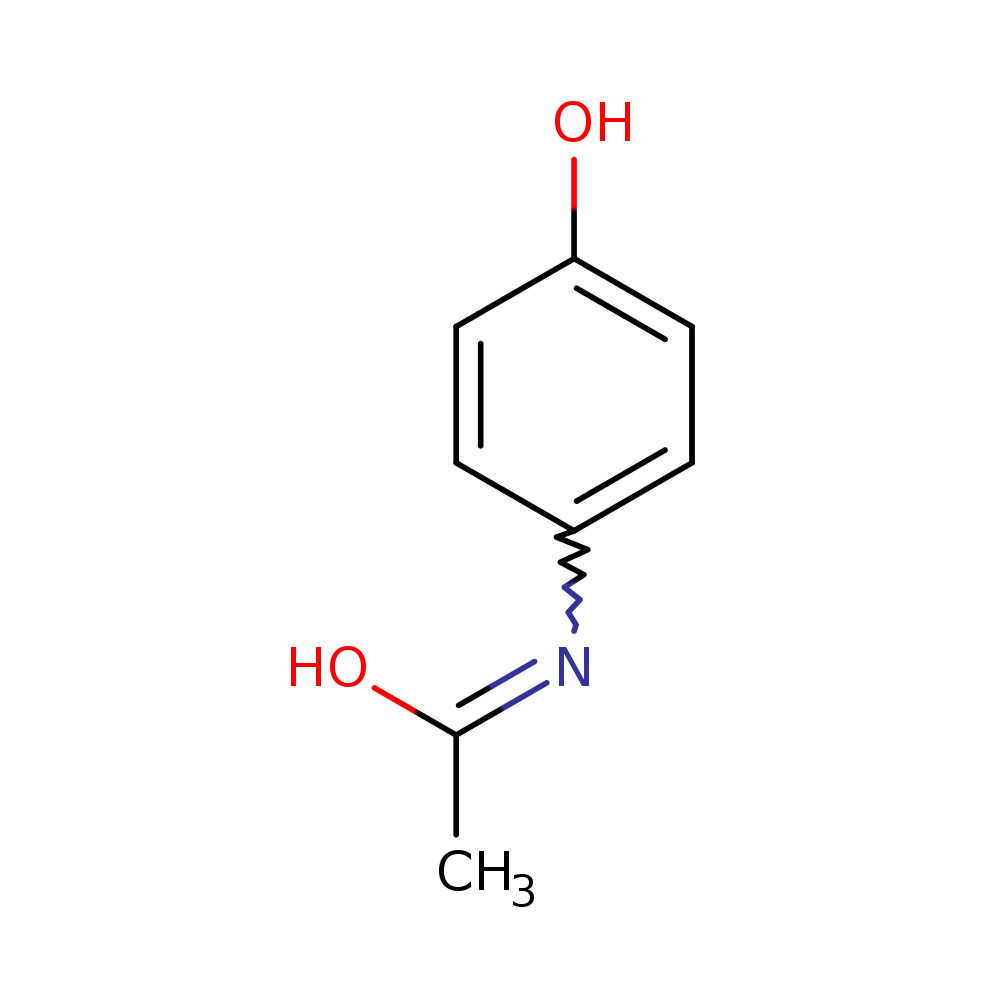
Alongside, the system identified the chemical formula associated with the peak as C8H9NO2. That is exactly the chemical formula for acetaminophen. It also identifies chemical structures below (the rightmost structure is acetaminophen). The pathway analysis we have in place identifies the acetaminophen structure as the most likely one amongst these candidates.
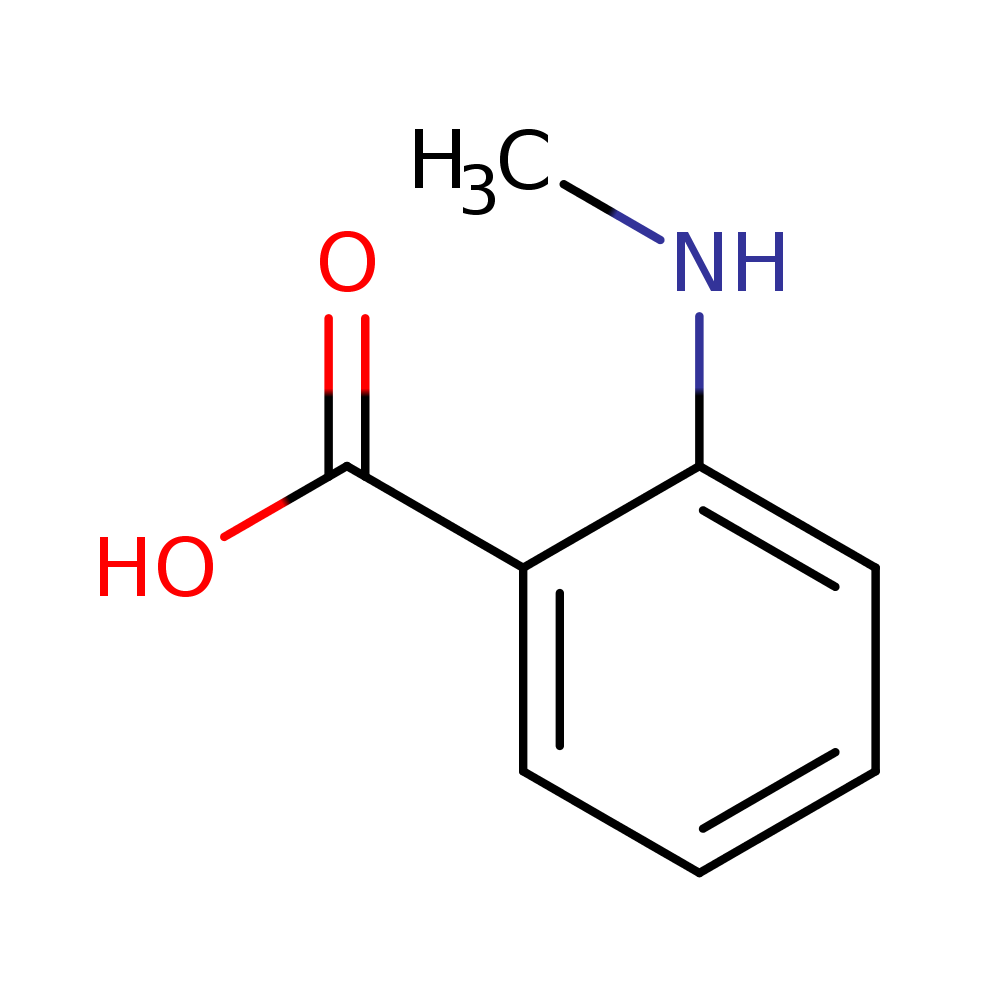
What gives us confidence in debugging engineered cells is the unbiased, untargeted, nature of this analysis. It highlights the major differences in the engineered cell versus the controls, and accurately predicts the chemical corresponding to it. Additional major identified peaks give visibility into side-effect changes, which would have been false negatives missed by a targeted analysis that was looking for specific products. We have been doing exactly this kind of targeted analysis for the last year, but can now recognize how much data we've been missing: all of these additional peaks are side effects of us putting a few genes in the cell, and all of these differences need to be taken into consideration when modifying the cellular chemistry further.
This untargeted (unbiased) analysis of every change in the cell gives us our debugging tool for biology, and deep learning has been the key to getting here reliably.
The debugging pipeline for biochemistry consists of several steps:
Let's concentrate on the deep learning module. If we were to plot the tuples (MZ, Retention Time, Intensity) from an LCMS trace, we get a 3D surface like the one below (left), or the heatmap below (right). Both images link to high resolution PDFs. Zooming into the heatmap to locate high intensity (red) peaks gives visibility into the density of the data.
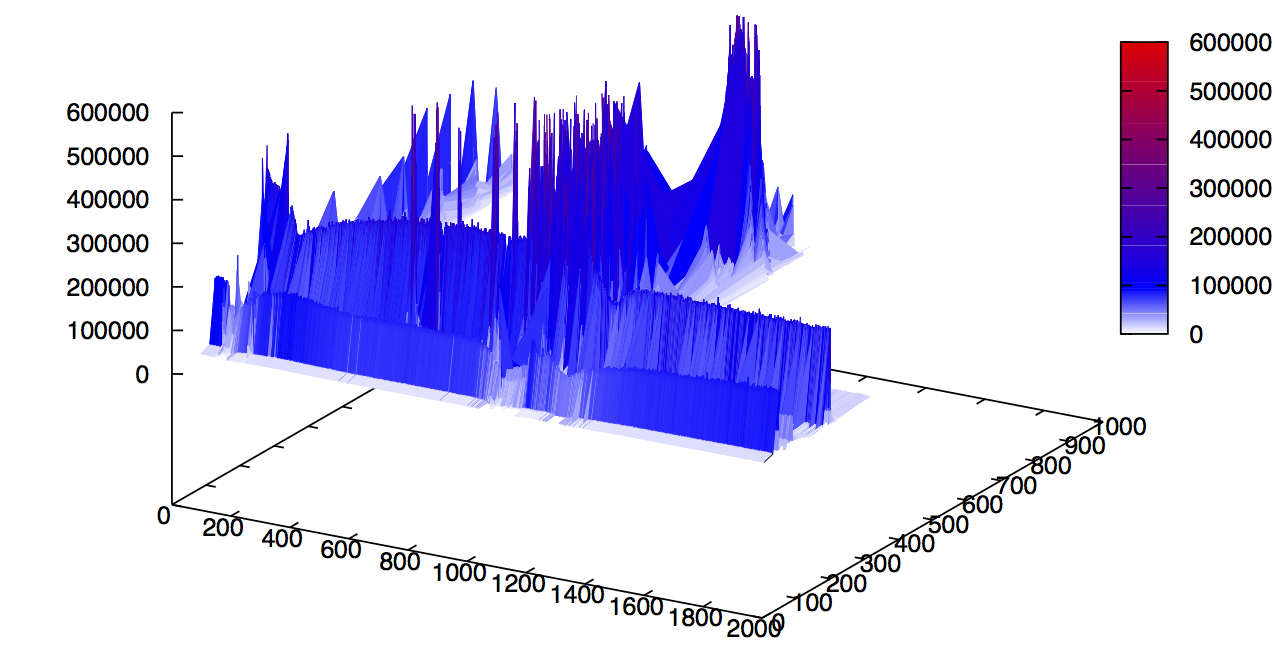
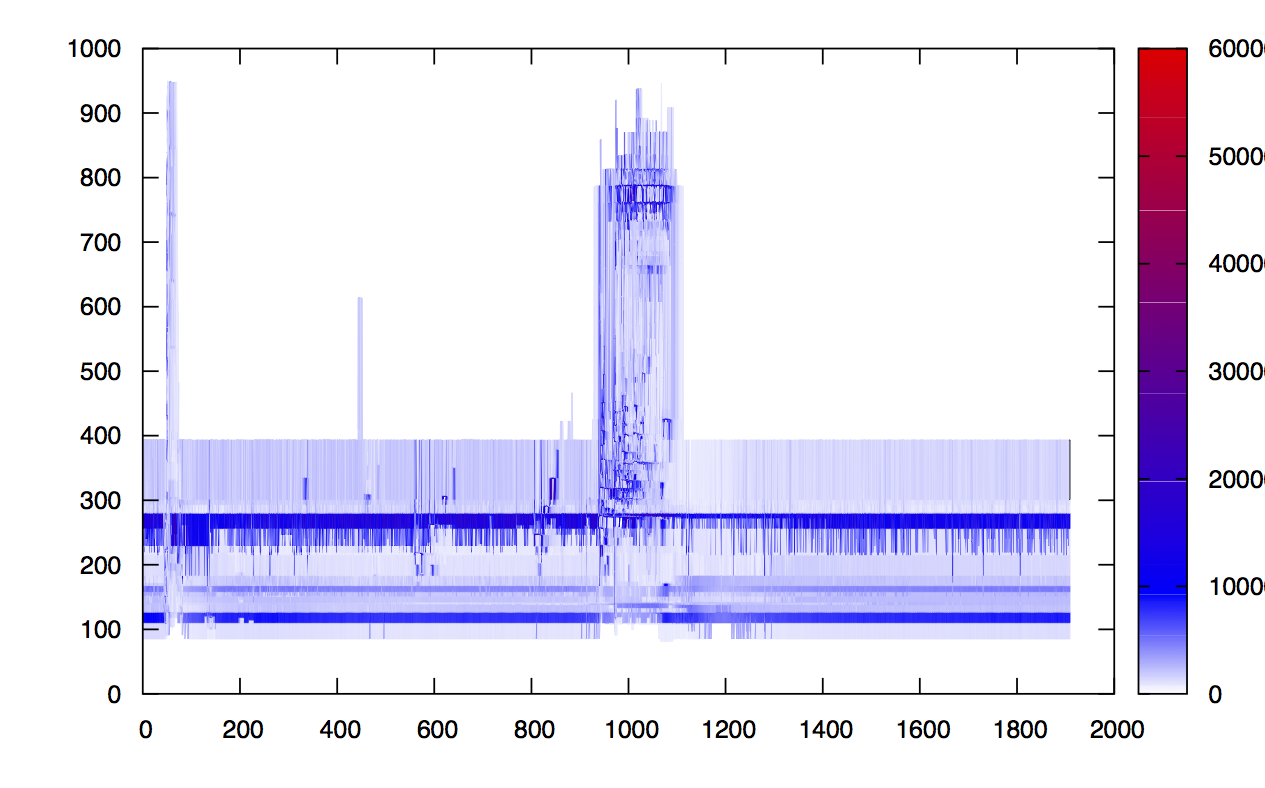
The global view has an overwhelming amount of data, so we look at a small window of about 50 data points, i.e., 0.0000025% of the data. Looking at the local 3D windows below, try to make an educated guess whether it is a "peak" or not.
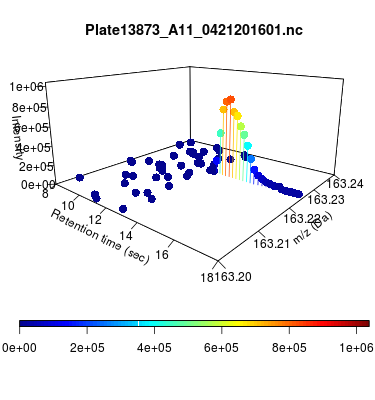
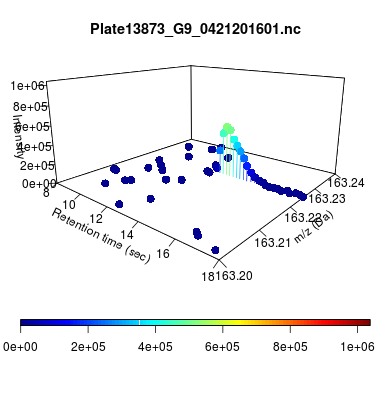
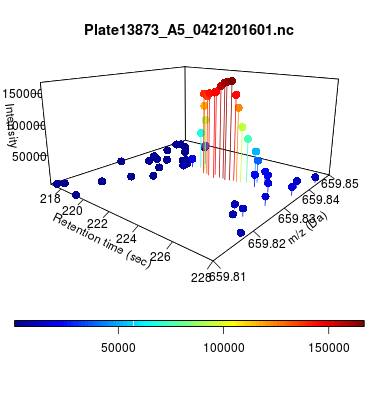
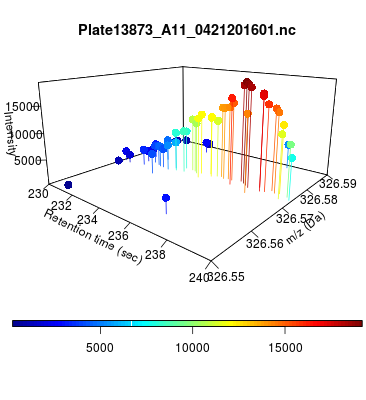
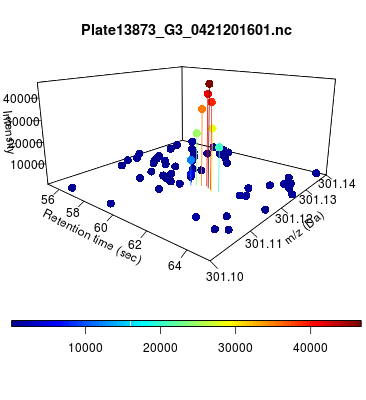
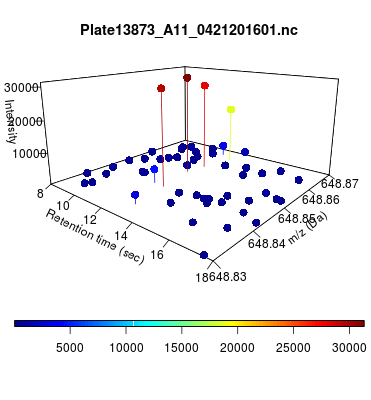
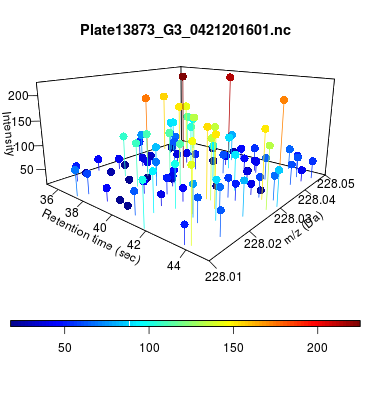
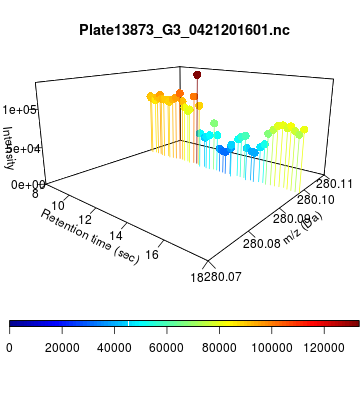
You probably called each one correctly. When we were initially analyzing these traces, we made manual visual assessments of peaks classifying them as "definitely is a peak," "definitely not a peak," and "maybe a peak."
While humans can easily recognize LCMS peaks by sight, developing a robust computational way to classify peaks can be difficult. We used deep learning to reduce the space down to a few dozen images, which can then be generalized across thousands of LCMS traces. Effectively, we used it to reduce the variation and complexity of LCMS data down to features that are common across traces.
Our first iteration used a trivial network. We found this to be marginally successful, but settled on using a deeper, multi-layer network which is better able to learn the nuances of LCMS traces. With this, we were able to successfully identify many peaks across a trace, while also excluding many peak-like features that a human would normally discard. Below, left is a group of peaks learnt by the network (which is easily seen to be valid peaks), while below right are features identified as noise.
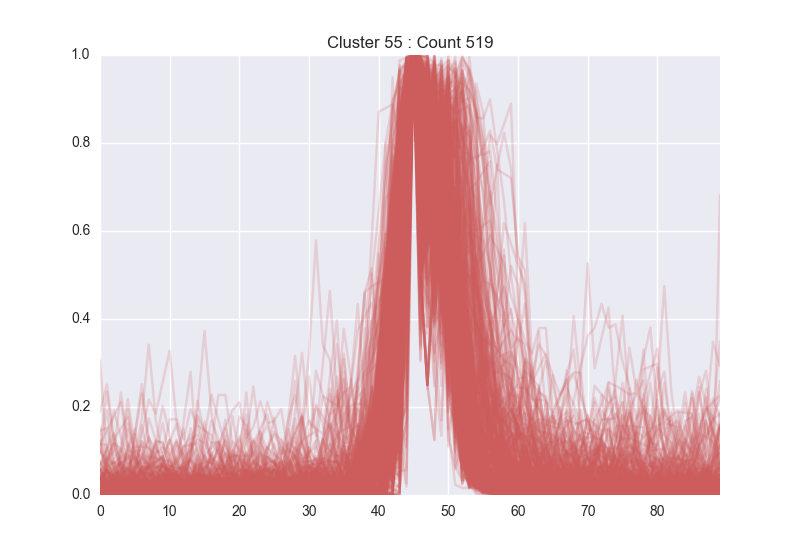
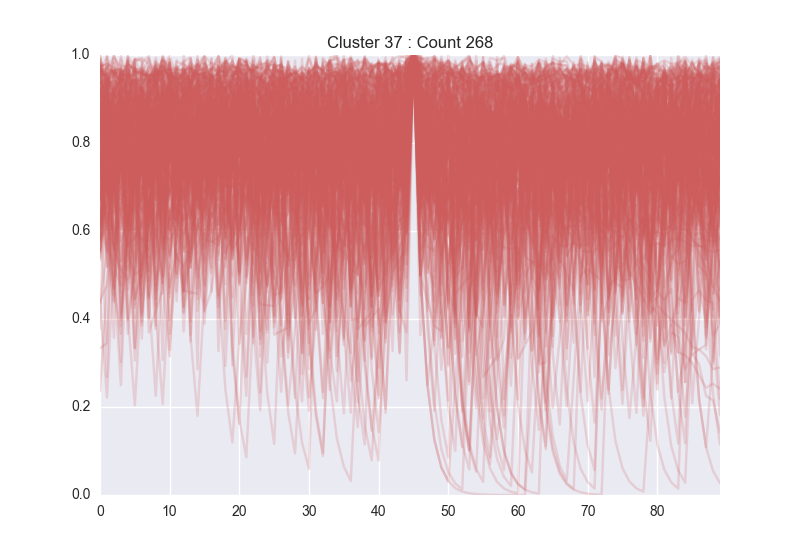
After showing that we could define individual peaks, we worked to generalize this method for doing large-scale differential analysis, which allows scientists to supply an experimental and control group and retrieve any molecule that is over- or under-expressed across their conditions.
Deep learning identifies differentials robustly. We build on that to extend our analysis to genetic changes across the compared cell lines. The entire pipeline brings together deep learning with other components. A customized biochemically aware SAT solver solves for chemical formulae. Network analysis that understands enzyme mechanisms, substrate specificity, and cellular connections derives molecular structures. Bioinformatics modules make the final mapping to genetic changes that explain the traces.




